本文介绍由兰州大学黎育权和腾讯量子实验室谢昌谕博士等人发表在Nature Machine Intelligence期刊上的研究成果,论文通讯作者为姚小军教授。文章中报道了一种自动图学习方法,能够在人工不参与的情况下,在多种不同任务上取得先进的预测性能,超越过去的主流模型。作者还提出一种新的分子鲁棒性实验方法,并发现模型集成能够大幅提升鲁棒性。

药物发现是一个耗时、昂贵和复杂的过程,在人类健康和福祉中发挥着至关重要的作用。机器学习方法,尤其是图学习方法有可能显著提高药物发现效率。它们能从现有药物相关数据集中进行学习,从而来预测分子相互作用和性质。同时,这也是机器学习能够从广阔的化学空间中以极快的速度和低成本寻找潜在候选药物的关键所在。
然而,在少部分数据集上追求高预测性能已经固化了它们的架构和超参数,这使得它们在新数据的利用上不在有优势。这种固化限制了他们在新数据上的学习能力和应用,使得他们的性能变得平庸。此外,大多数图学习方法严重依赖深度学习的专家知识来实现其声称的最先进结果。当作者不给出这些具体的神经网络架构和参数的时候,后来的研究人员甚至无法复现他们的模型性能。
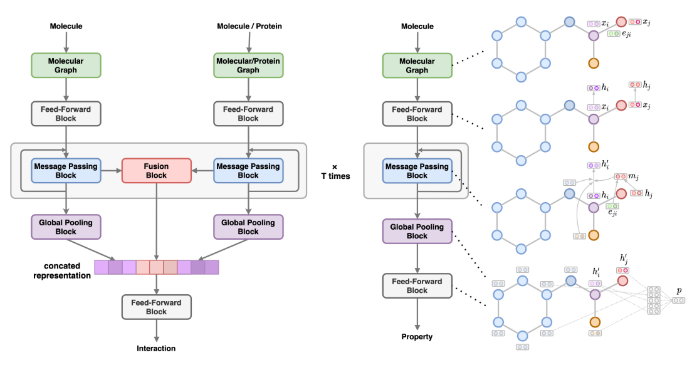
这项工作提出了基于图学习的自适应机器(Graph learning based adaptive machine, GLAM),它可以适应数据集并在无需人工干预的情况下做出准确的预测。
(1)提出一种自动机器学习方法,可以同时应用于分子属性预测、药物-蛋白相关关系预测、药物-药物相互作用预测。
(2)在相对公平比较的情况下(相同数据集分割),对比目前主流模型并取得最佳性能,包括分子属性预测任务,药物-靶标相关关系预测任务。
(3)提出一种合理的图模型鲁棒性测试方法,并发现集成模型比单模型的鲁棒性好。
相关文章

